Acute coronary syndrome (ACS) is one of the most severe subtypes of coronary heart disease (CHD), mainly including acute myocardial infraction (AMI) and unstable angina pectoris (UAP). Despite certain progress in the aspects of prevention, diagnosis and treatment, ACS remains one of the major causes of mortality in China and worldwide. To date, genome-wide association studies (GWAS) have identified multiple CHD-associated genetic variations but are unable to explain the impacts of environment factors on diseases. The concrete regulatory mechanism remains unclear. The DNA methylation (DNAm) is a reversible epigenetic modification, which integrates environmental factors and genetic susceptibility to regulate gene expression without altering DNA sequence, thus has gained increasing research interests in exploring pathogenesis of ACS. DNAm profile reflects long-term individual lifestyle and environmental exposure and provides more dynamic disease risk information than genetic variation. A growing body of evidence supported the crucial link between DNAm alteration and ACS. However, most previously published studies could not distinguish the direction of causality between DNAm changes and ACS due to their cross-sectional design. Thus, it is of great importance to understand the effects of DNAm alteration related to ACS in uncovering disease pathogenesis, discovering novel risk prediction biomarkers, and formulating individual strategies for prevention and treatment.
On August 28, 2024, Tangchun Wu and Chaolong Wang from Huazhong University of Science and Technology and Jun Lv from Peking Univeristy are joint corresponding authors of an online published research article, entitled “Genome-wide DNA methylation profiling in blood reveals epigenetic signature of incident acute coronary syndrome” on Nature Communications. This research conducted an epigenome-wide association study (EWAS) based on two prospective cohorts in China, Dongfeng-Tongji (DFTJ) cohort and China Kadoorie Biobank (CKB) and integrated multi-omics data including genome, epigenome, and transcriptome to reveal the key impact of DNAm alteration in pathogenetic process of ACS and the superior disease risk prediction ability. Post-doctor Pinpin Long from Huazhong University of Science and Technology and Dr. Jiahui Si from Peking University are co-first authors of this article.
Key points
First, researchers conducted a two-stage EWAS design based on two independent prospective cohort in China. In the discovery stage, researchers conducted EWAS analysis covering more than 770,000 CpGs based on 751 pairs of ACS cases and matched controls nested in DFTJ cohort and identified 72 differentially methylated positions (DMPs) associated with incident ACS (FDR < 0.05). In the replication stage, a total of 26 DMPs showed directionally consistent and statistically significant relationships among nested case-control study of incident ACS in CKB (476 pairs) (Figure 1). Three of the 26 DMPs were mapped to known cardiovascular disease (CVD) susceptibility genes, including PRKCZ, PRDM16, and EHBP1L1. This study further uncovered the epigenetic regulatory mechanism of identified DMPs during the development of ACS. Mendelian randomization (MR) analysis further verified the causal relationship between four DMPs (mapped to PRKCZ, TRIM27, EMC2, and EHBP1L1) and ACS.
Then, researchers evaluated the potential regulatory roles of validated DMPs on gene expression by expression quantitative trait methylation analysis (eQTM) and found that cg03609847 mapped to PIGG, cg12853539 mapped to HDDC2, and cg16749093 mapped to EHBP1L1 were hypomethylated among incident ACS group and were significantly and negatively correlated with their mapped genes. Gene expression dataset of multiple tissues further pinpointed that the PIGG and EHBP1L1 were significantly up-regulated in leukocytes among ACS patients and showed significantly higher expression levels in carotid atheroma plaques than normal carotid tissues or in advanced atherosclerotic carotid artery segments than early atherosclerotic carotid segments (Figure 2 & 3). These consistent results further supported that the potential gene regulatory functions of the specific DMPs the pathogenesis of ACS and provided solid scientific evidence of these CpGs served as novel preventive and therapeutic targets.
Last, researchers evaluated the clinical application value of identified DMPs in the prediction of incident ACS and constructed methylation risk score (MRS) based on 67 DMPs. Results indicated that MRS had better predictive performance in disease risk prediction than traditional risk factors and polygenic risk scores (PRSs) in both cohorts, which further proved the substantial capability of DNAm in disease risk prediction (Figure 4).
Conclusion and prospect
This study elucidated the pivotal role of DNAm in pathogenesis of ACS and presented its important application potential in disease risk prediction through large-scale two-stage EWAS and systematic multi-omics integration analysis. These findings provided novel epigenetic evidence and highlighted the possibility of DNAm as potential biomarkers in prediction, intervention, and treatment. Besides, constructed MRS based on DMPs showed remarkable clinical application value in ACS risk prediction. Further studies in other populations and animal models are warranted to ensure the generalizability of those findings and uncover the in-depth biological mechanism in order to provide promising evidence for personalized medicine and early intervention strategies.
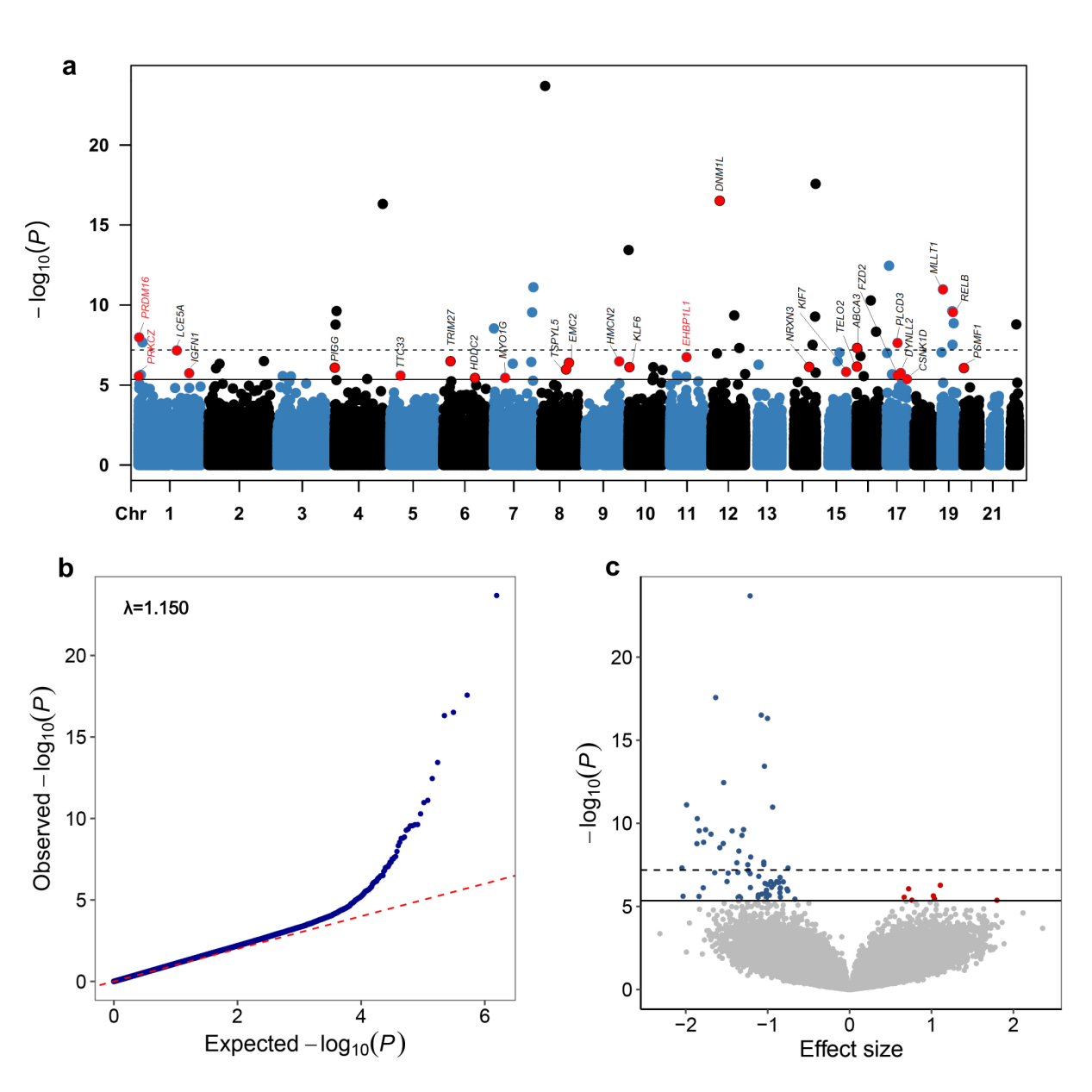
Figure 1. Epigenome-wide association study of incident ACS in the Dong-feng Tongji cohort.
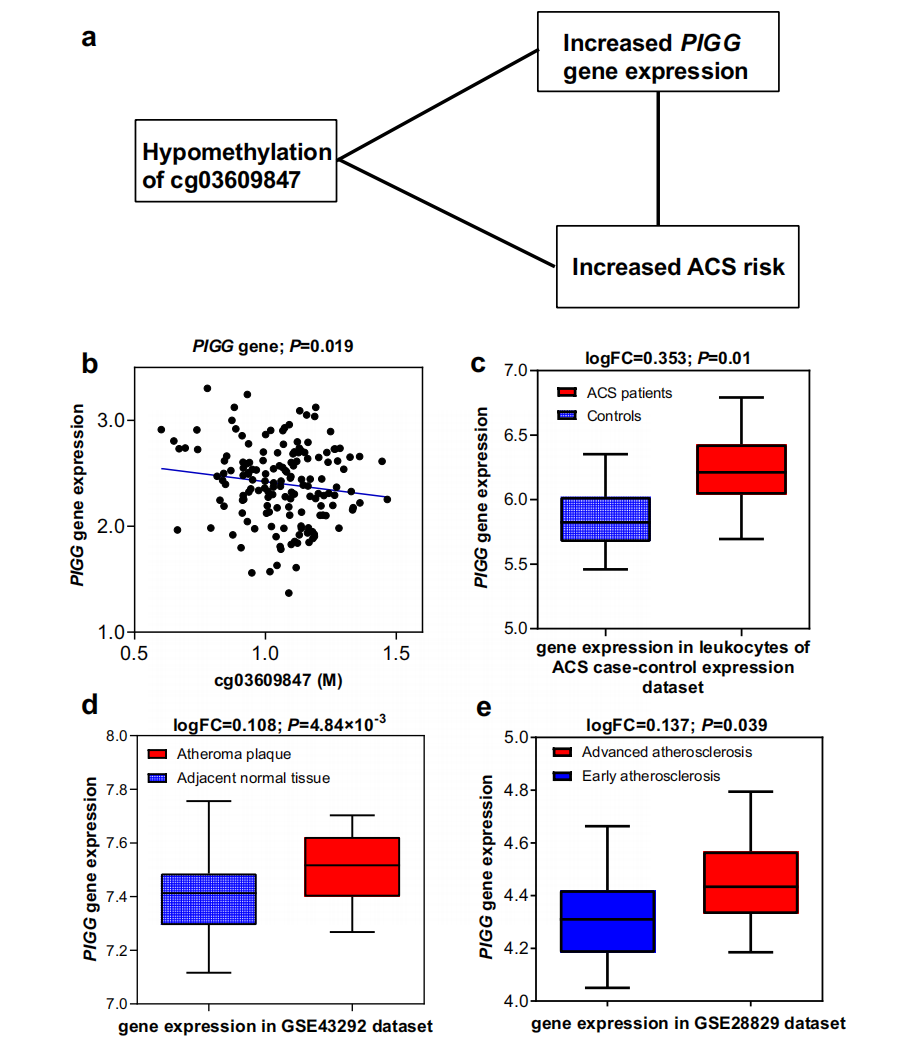
Figure 2. Three-way association among cg03609847, PIGG gene expression, and ACS risk.
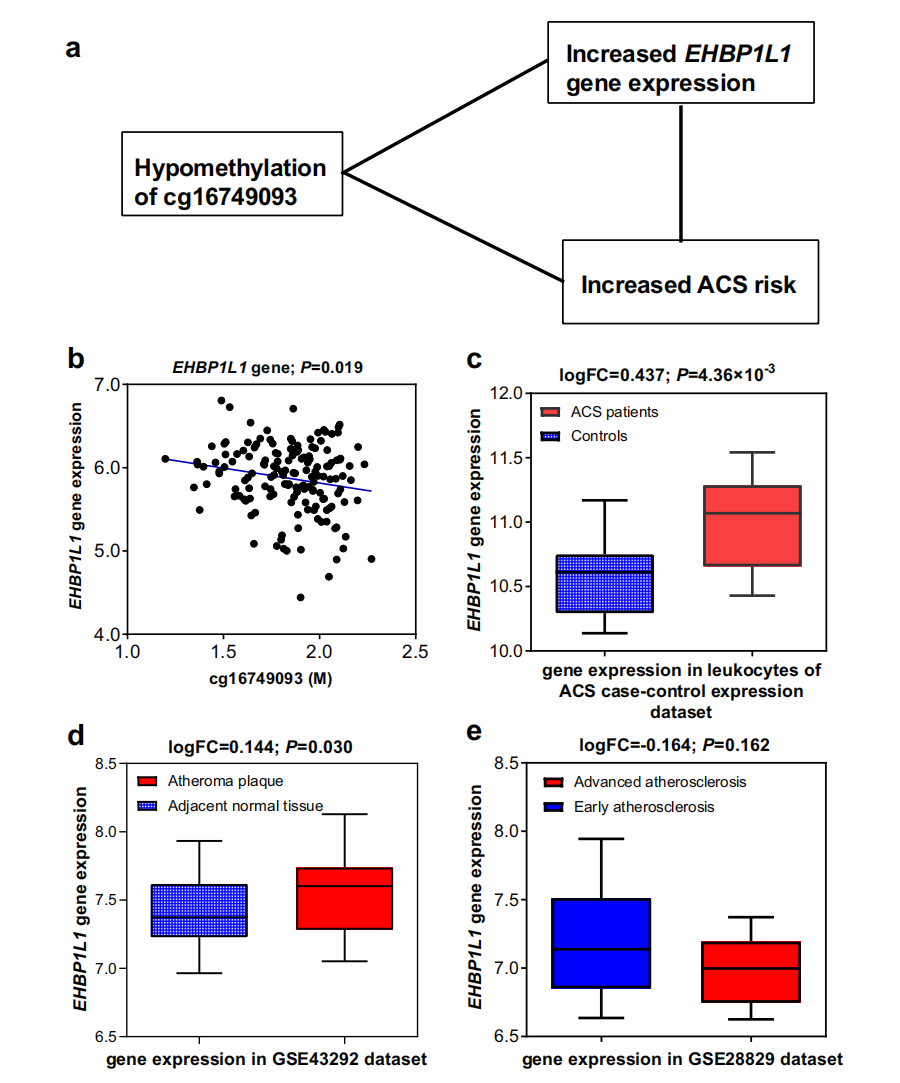
Figure 3. Three-way association among cg16749093, EHBP1L1 gene expression, and ACS risk.
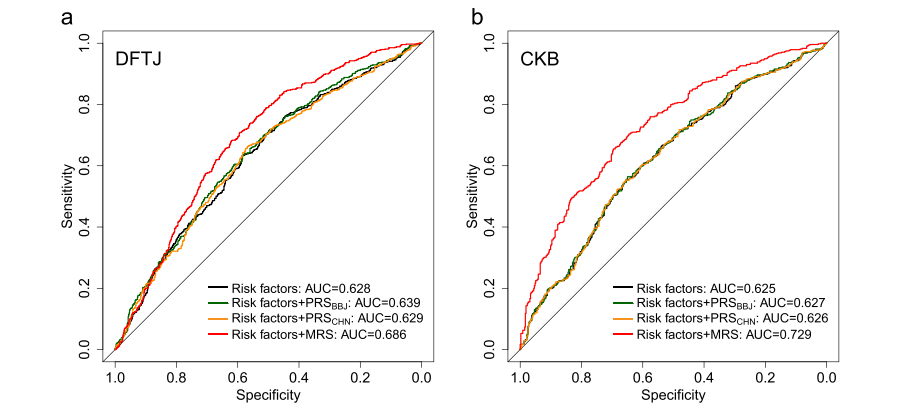
Figure 4. Evaluation of MRS, PRS, and traditional risk factors in the prediction of incident ACS

